


Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical DevicesMedical deviceThe Pharmaceutical Machinery Law has been amended by the original Pharmaceutical Law, and its specific implementation provisions include the Pharmaceutical Machinery Law Implementation Order, the QMS Provincial Order and other relevant announcements and notices.
b) Competent authorities
Japan’s drug and medical device management is mainly administered by two competent authorities, namely the Japanese Ministry of Health and Labour (MHLW) and the Administrative Authority for Medical and Medical Devices (PMDA).
Classification of Medical Devices
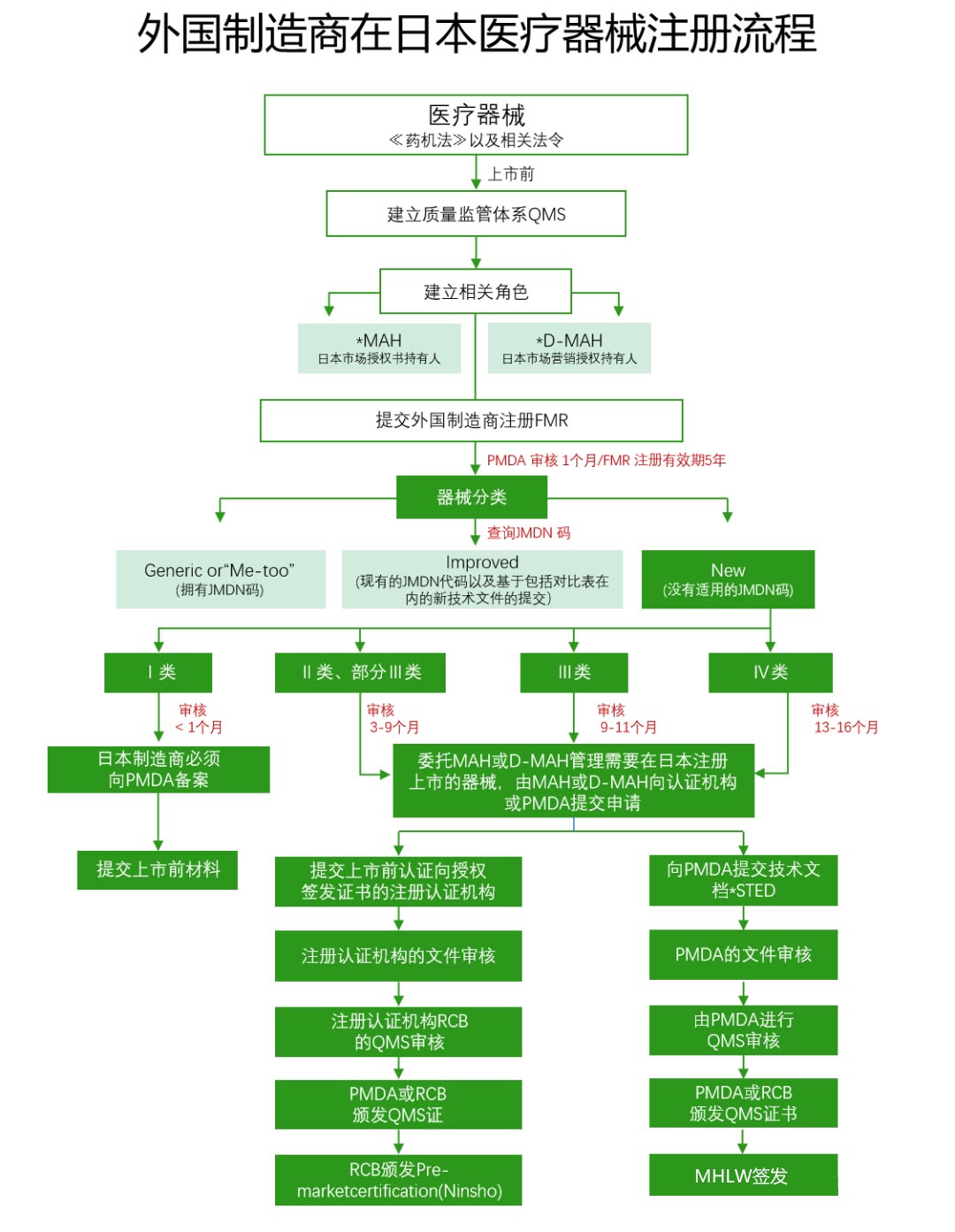
General Medical Devices (Class I)Designated by the Ministry of Health and Labour of Japan after hearing the opinion of the Pharmaceutical and Food Hygiene Review Board, such medical devices have a lower risk of human life and health impacts when side effects or functional impairment occur.
Management of Medical Devices (Class II):Also designated by the Ministry of Health and Labour of Japan after hearing the opinion of the Pharmaceutical and Food Hygiene Review Board, such medical devices may affect human life and health when side effects or functional impairment occur, and therefore need to be properly managed.
Highly Managed Medical Devices (Class III and Class IV):Such medical devices can have serious consequences for human life and health in the event of side effects or functional impairment, so strict management must be carried out.

Medical Device Certification Model and Registration Audit Process
Foreign manufacturers intending to export their equipment to Japan must register with the Ministry of Health, Labour and Welfare, a process known as Foreign Manufacturer Registration (FMR). This procedure follows the PMD Act & Order No. 169 ISO 13485 Japanese medical device registration review process.
V. Focus on concerns
Market Authorization Holders (MAH) and Marketing Authorization Holders (D-MAH)Foreign manufacturers must designate market authorization holders in Japan, which is the first condition for selling medical devices in Japan.
2) STED summary (registered information):The requirements include product specifications, stability and shelf life data, performance test data, risk analysis, and clinical data.
Audit of Quality Management System (QMS):Executed by the Pharmaceutical and Medical Device Administration (PMDA) or a registered certification body (RCB), the scope of the audit includes all relevant locations such as manufacturing vendors, medical device design, manufacturing and manufacturing.
Multilateral regulatory requirements:Imported medical devices to the Japanese market also need to meet the requirements of other regulations, such as the Electrical Safety Law, the Radiowave Law, etc.
 Follow the customer service WeChat account.
Follow the customer service WeChat account.